Vertical integration for paired datasets
uniPort also involves a vertical integration method for paired-cell datasets. Here we use uniPort to integrates paired SNARE-seq CellLineMixture datasets from GSE126074.
[1]:
import uniport as up
import numpy as np
import pandas as pd
import scanpy as sc
import episcanpy as epi
from sklearn.preprocessing import MinMaxScaler
[2]:
labels = pd.read_csv('snare/cell_line_meta.txt', sep='\t')
celltype = labels['cell_line'].values
[3]:
adata_peaks = up.load_file('snare/GSE126074_CellLineMixture_SNAREseq_chromatin_counts.tsv')
adata_rna = up.load_file('snare/GSE126074_CellLineMixture_SNAREseq_cDNA_counts.tsv')
print(adata_peaks)
print(adata_rna)
AnnData object with n_obs × n_vars = 1047 × 136771
AnnData object with n_obs × n_vars = 1047 × 18666
[4]:
adata_peaks.obs['cell_type'] = celltype
adata_peaks.obs['domain_id'] = 0
adata_peaks.obs['domain_id'] = adata_peaks.obs['domain_id'].astype('category')
adata_peaks.obs['source'] = 'ATAC'
adata_rna.obs['cell_type'] = celltype
adata_rna.obs['domain_id'] = 1
adata_rna.obs['domain_id'] = adata_rna.obs['domain_id'].astype('category')
adata_rna.obs['source'] = 'RNA'
Preprocess scATAC-seq peaks. Select 2,000 highly variable peaks.
[5]:
adata_peaks.X[adata_peaks.X>1] = 1
epi.pp.select_var_feature(adata_peaks, nb_features=2000, show=False, copy=False)
sc.pp.normalize_total(adata_peaks)
up.batch_scale(adata_peaks)
print(adata_peaks)
AnnData object with n_obs × n_vars = 1047 × 2070
obs: 'cell_type', 'domain_id', 'source'
var: 'n_cells', 'prop_shared_cells', 'variability_score'
Preprocess scRNA-seq peaks. Select 2,000 highly variable genes.
[6]:
sc.pp.normalize_total(adata_rna)
sc.pp.log1p(adata_rna)
sc.pp.highly_variable_genes(adata_rna, n_top_genes=2000, inplace=False, subset=True)
up.batch_scale(adata_rna)
print(adata_rna)
AnnData object with n_obs × n_vars = 1047 × 2000
obs: 'cell_type', 'domain_id', 'source'
var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
Project scATAC-seq into latent space with the help of scRNA-seq. The latent representations of scATAC-seq are stored at adata_atac.obs['latent']
[7]:
adata_peaks = up.Run(adatas=[adata_peaks, adata_rna], mode='v', lr=0.001, iteration=10000)
Dataset 0: ATAC
AnnData object with n_obs × n_vars = 1047 × 2070
obs: 'cell_type', 'domain_id', 'source'
var: 'n_cells', 'prop_shared_cells', 'variability_score'
Dataset 1: RNA
AnnData object with n_obs × n_vars = 1047 × 2000
obs: 'cell_type', 'domain_id', 'source'
var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
Reference dataset is dataset 1
Epochs: 100%|████████████████████████████| 2500/2500 [16:26<00:00, 2.53it/s, recon_loss=239.627,kl_loss=7.374]
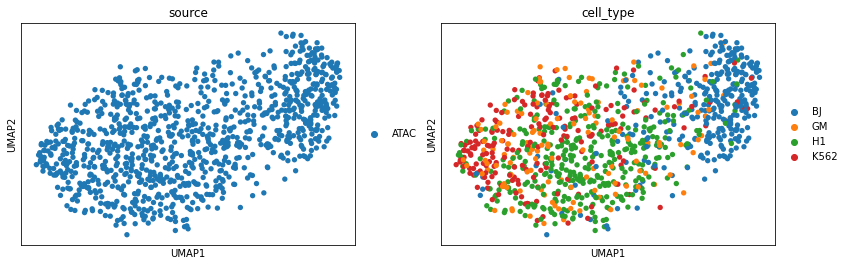
Perform UMAP visualization for scATAC before vertical integration.
[8]:
sc.pp.pca(adata_peaks)
sc.pp.neighbors(adata_peaks)
sc.tl.umap(adata_peaks, min_dist=0.1)
sc.pl.umap(adata_peaks, color=['source', 'cell_type'])
... storing 'cell_type' as categorical
... storing 'source' as categorical
Perform UMAP visualization for scRNA.
[9]:
sc.pp.pca(adata_rna)
sc.pp.neighbors(adata_rna)
sc.tl.umap(adata_rna, min_dist=0.1)
sc.pl.umap(adata_rna, color=['source', 'cell_type'])
... storing 'cell_type' as categorical
... storing 'source' as categorical

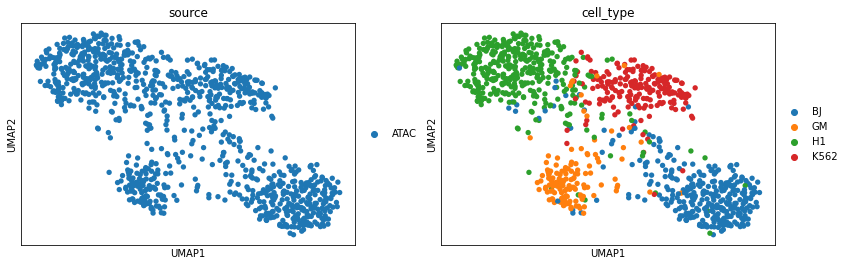
Perform UMAP visualization for scATAC latent representations after vertical integration. The cell types of scATAC-seq now are more distinguishable than before.
[10]:
sc.pp.neighbors(adata_peaks, use_rep='latent')
sc.tl.umap(adata_peaks, min_dist=0.1)
sc.pl.umap(adata_peaks, color=['source', 'cell_type'])
[ ]: