Home
Tutorials
- Home
- Installation
- Examples
- API and modules
- uniport.Run
- uniport.label_reweight
- uniport.load_file
- uniport.filter_data
- uniport.batch_scale
- uniport.data_loader.SingleCellDataset
- uniport.data_loader.SingleCellDataset_vertical
- uniport.data_loader.load_data
- uniport.model.vae.VAE
- uniport.model.layer.DSBatchNorm
- uniport.model.layer.Block
- uniport.model.layer.NN
- uniport.model.layer.Encoder
- uniport.model.layer.Decoder
- uniport.model.loss.kl_div
- uniport.model.loss.distance_matrix
- uniport.model.loss.distance_gmm
- uniport.model.loss.unbalanced_ot
- uniport.model.utils.onehot
- uniport.model.utils.EarlyStopping
- uniport.metrics.batch_entropy_mixing_score
- uniport.metrics.silhouette
- uniport.metrics.label_transfer
- uniport.logger.create_logger
- Release
![]()
![]()
The original paper: a unified single-cell data integration framework with optimal transport
Website and documentation: https://uniport.readthedocs.io
Source Code (MIT): https://github.com/caokai1073/uniport
All data before and after processing in Examples are available at source data link
Author’s Homepage: www.caokai.site
Installation
The uniport package can be installed via pip:
pip3 install uniport
Main function
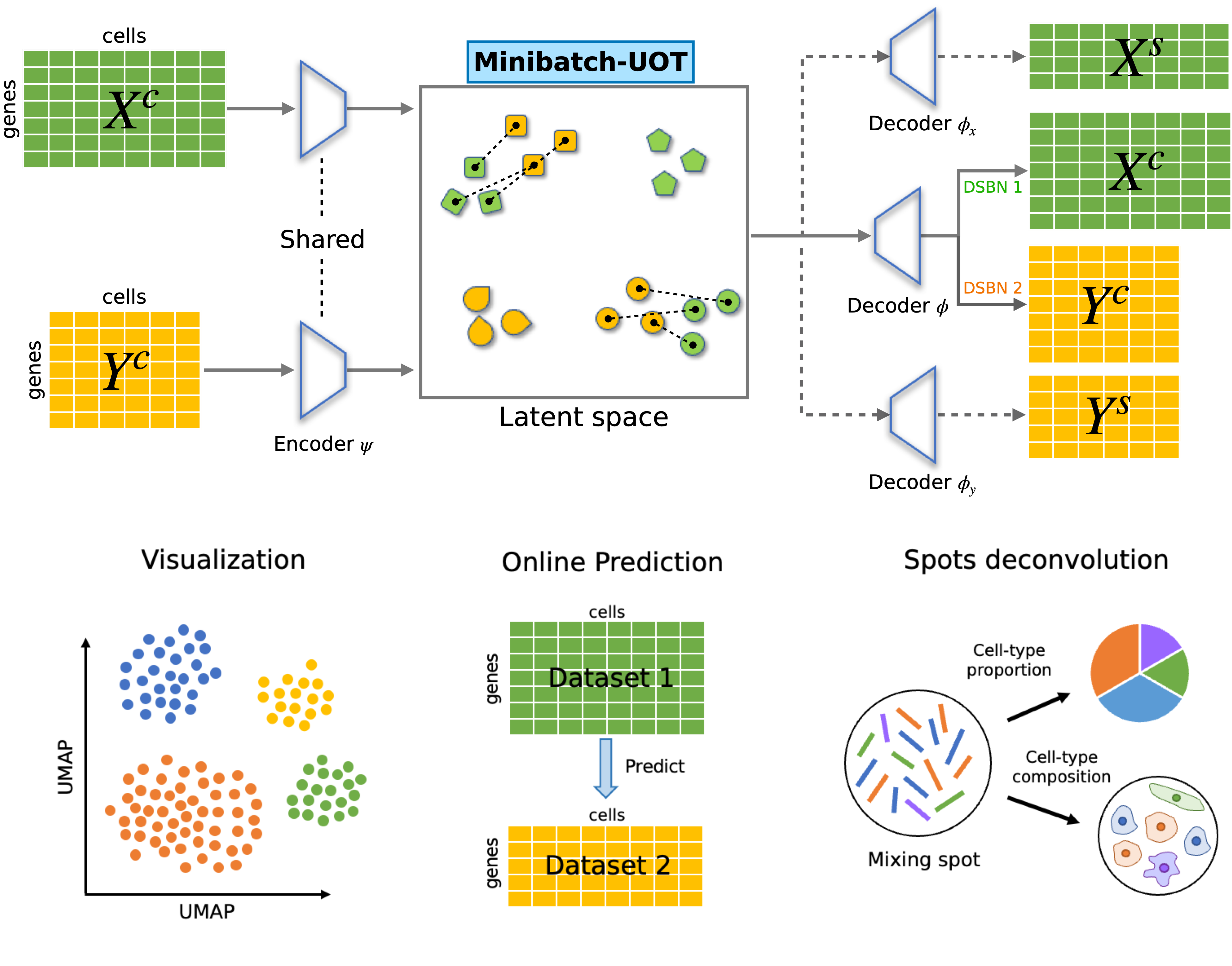
uniport.Run(…)
Key parameters includes:
adatas: List of AnnData matrices for each dataset.
adata_cm: AnnData matrix containing common genes from different datasets.
mode: Choose from [‘h’, ‘v’, ‘d’] If ‘mode=h’, integrate data with common genes (Horizontal integration). If ‘mode=v’, integrate data profiled from the same cells (Vertical integration). If ‘mode=d’, inetrgate data without common genes (Diagonal integration). Default: ‘h’.
lambda_s: balanced parameter for common and specific genes. Default: 0.5
lambda_recon: balanced parameter for reconstruct term. Default: 1.0
lambda_kl: balanced parameter for KL divergence. Default: 0.5
lambda_ot: balanced parameter for OT. Default: 1.0
iteration: max iterations for training. Training one batch_size samples is one iteration. Default: 30000
ref_id: id of reference dataset. Default: The domain_id of last dataset
save_OT: if True, output a global OT plan. Need more memory. Default: False
out: output of uniPort. Choose from [‘latent’, ‘project’, ‘predict’]. If out==’latent’, train the network and output cell embeddings. If out==’project’, project data into the latent space and output cell embeddings. If out==’predict’, project data into the latent space and output cell embeddings through a specified decoder. Default: ‘latent’
import uniport as up
import scanpy as sc
# HVG: highly variable genes
adata1 = sc.read_h5ad('adata1.h5ad') # preprocessed data with data1 specific HVG
adata2 = sc.read_h5ad('adata2.h5ad') # preprocessed data with data2 specific HVG, as reference data
adata_cm = sc.read_h5ad('adata_cm.h5ad') # preprocesssed data with common HVG
# integration with both common and dataset-specific genes
adata = up.Run(adatas=[adata1, adata2], adata_cm=adata_cm)
# save global optimal transport matrix
adata, OT = up.Run(adatas=[adata1, adata2], adata_cm=adata_cm, save_OT=True)
# integration with only common genes
adata = up.Run(adata_cm=adata_cm)
# integration without common genes
adata = up.Run(adatas=[adata1, adata2], mode='d')
# integration with paired datasets
adata = up.Run(adatas=[adata1, adata2], mode='v')
Citation
@article{Cao2022.02.14.480323,
author = {Cao, Kai and Gong, Qiyu and Hong, Yiguang and Wan, Lin},
title = {uniPort: a unified computational framework for single-cell data integration with optimal transport},
year = {2022},
doi = {10.1101/2022.02.14.480323},
publisher = {Cold Spring Harbor Laboratory},
journal = {bioRxiv}}